




文章标题:Applying multi‐omics toward tumor microbiome research
发表期刊:iMeta
作者单位:俄亥俄州立大学医学院放射肿瘤学系、俄亥俄州立大学詹姆斯综合癌症中心癌症代谢中心、俄亥俄州立大学医学院生物化学和药理学系肿瘤微生物在肿瘤发生、发展、转移和治疗过程中起着重要作用,且极有可能成为肿瘤早期诊断和治疗的标志物,但同时也存在一定的挑战,如肿瘤内微生物含量低和不可培养,因此亟需新的高分辨率的技术来支持后续的研究。多组学技术(基因组学,转录组学,蛋白质组学和代谢组学)能从不同层次阐述肿瘤微生物,将对未来微生物与肿瘤微环境相互作用的研究产生巨大影响。本文系统总结了多组学技术的研究进展及其在肿瘤微生物组研究中的应用,为研究者提供了一个可供参考的组学工具箱。
1.引言
肿瘤微环境(Tumor Microenvironment, TME)中存在微生物并受其影响,目前地球上大约有1012种已知的微生物,只有11种被国际癌症研究机构(International Agency for Research on Cancer, IARC)确认为“肿瘤微生物”。研究人员发现微生物活体存在于多种肿瘤中(乳腺癌、结直肠癌、肝细胞癌、胰腺癌、皮肤癌、肾细胞癌,胃癌、肺癌、前列腺癌和鼻咽癌等;图1),而且不同类型肿瘤中的微生物有惊人的相似之处。肿瘤内微生物通过DNA损伤、致癌激活途径、抗肿瘤药物分解代谢和免疫系统调节等多种机制影响肿瘤的发展和治疗(如:大肠杆菌释放能够诱导致癌DNA突变的基因毒素;幽门螺杆菌通过诱导炎症和影响调节粘膜细胞生长和增殖的关键细胞内信号通路;用于治疗胰腺导管腺癌的化疗药物吉西他滨被肿瘤内细菌代谢至失活状态,从而导致耐药性)。
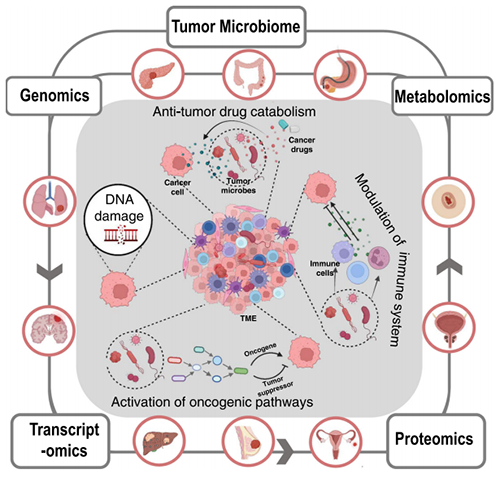
图1. 微生物的致癌作用以及多组学方法研究肿瘤微生物
2.用于肿瘤微生物组研究的基因组学和转录组学
基因组学和转录组学专注于研究参与基因表达调控的核酸(DNA和RNA)。目前,使用最广泛的测序技术(图2)包括三种二代测序系统(即Illumina、Ion Torrent、BGI)和两种三代测序技术(即PacBio和纳米孔)。这些测序方法(DNA测序,RNA测序,16S rRNA测序,表观遗传学测序(如ChIP-seq和DNA/RNA甲基化测序)和三维基因组技术)是发展各种基因组和转录组研究方法的基础。此外,基因编辑工具的最新研究进展已经能够在全基因组水平上进行多基因操作。
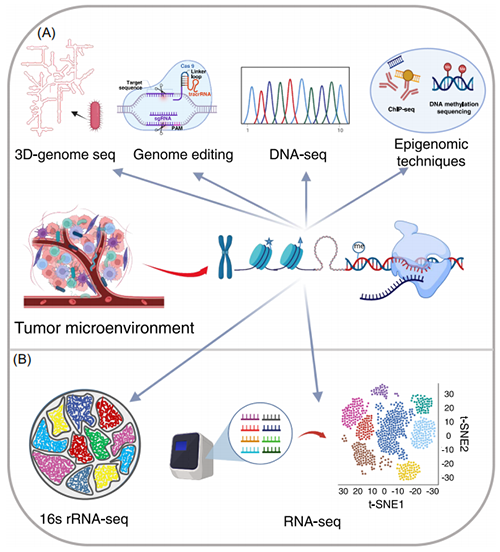
图2. 用于肿瘤微环境研究的基因组和转录组研究技术
2.1 DNA测序
DNA-seq通常用于三种类型的基因组分析:全基因组测序(Whole Genome Sequencing, WGS)、全外显子测序(whole exome sequencing, WES)和靶向测序。随着DNA技术的快速发展,DNA-seq正成为研究不可培养微生物(包括人类微生物组中的微生物)的有力工具。基因组文库的测序和异源表达极大地促进了我们对代谢途径和肿瘤微生物生理的理解。
2.2 16S rRNA测序
16S rRNA测序是一种专门针对细菌和古细菌表达的30S核糖体亚单位16S的测序技术;该序列包含交替的高度保守和高变区;保守区可用于设计扩增靶向片段的通用引物,而高变区的分析可用于鉴定特定的细菌物种,尽管一些注释可能低于物种水平。在肿瘤微生物组领域,16S rRNA测序主要用于研究群落中物种的组成、物种间的进化关系以及微生物群落多样性。
2.3 RNA测序(RNA-seq)
RNA-seq是一种转录组测序方法,其中高通量测序方法用于研究复杂样品中不同RNA的群体,包括信使RNA(messenger RNA, mRNA)、转运RNA(trainsfer RNA, tRNA)、非编码RNA(noncoding RNA, ncRNA)和微小MicroRNA(micro RNA, miRNA)等。在过去几十年里,RNA-seq技术已成为在转录组水平上分析基因表达不可或缺的工具。在RNA生物学领域,RNA-seq已广泛应用于研究单细胞基因转录、蛋白质表达和RNA结构。RNA-seq技术的进步也进一步促进了空间转录组学的出现和发展;直接长阅读或全长RNA-seq方法、以及不断更新的数据分析方法,使研究人员能够更好地了解RNA生物学(包括转录过程机制以及折叠和分子间相互作用对RNA功能的影响)。总的来说,RNA-seq是通过基因转录水平上的分析来研究癌细胞和肿瘤微生物组通路交互影响的强有力工具。
2.4 表观遗传学技术
表观遗传学研究不涉及DNA序列改变的表型变化。与遗传调节不同,典型的表观遗传变化(包括DNA/RNA修饰和组蛋白翻译后修饰)是可逆的,且不影响细胞基因组序列。在真核细胞中,核小体作为染色质组织的基本重复单位,主要由围绕核心组蛋白八聚体缠绕1.5次的147个碱基对组成(如两拷贝H2A、H2B、H3和H4)。DNA、RNA和组蛋白的共修饰是调控特定基因激活和抑制的关键机制。表观遗传调控在研究与人类疾病特别是癌症密切相关的DNA复制、转录和损伤修复过程中起重要的调控作用。
DNA甲基化在不改变基因序列的情况下调控转录,而肿瘤抑制相关基因表达的关闭会导致肿瘤的发生。大量研究表明DNA异常甲基化与多种肿瘤的发生发展密切相关,因而DNA甲基化水平的改变常被检测为肿瘤诊断的标志。目前,已经开发了多种DNA甲基化测序技术,如全基因组亚硫酸氢盐测序(Whole Genome Bisulfite Sequencing, WGBS),简化的亚硫酸氢盐测序(Reduced representation bisulfite sequencing, RRBS),氧化亚硫酸氢盐测序(oxBS-seq)和无细胞甲基化DNA免疫沉淀和高通量测序(cfMeDIP-seq)。
组蛋白翻译后修饰(Post-translational Modifications, PTMs)包括甲基化、磷酸化、乙酰化、巴豆酰化、泛素化、糖基化、糖化和ADP核糖基化,不同组蛋白PTMs被认为与癌症的发生发展有关,如微生物的甲基乙二醛合酶(methylglyoxal synthases, MGSs)可以促进TME中甲基乙二醛的生成;组蛋白MGO(methylglyoxal)-糖基化被发现通过影响染色质的三维结构影响肿瘤的发展。
此外,染色质免疫沉淀测序(Chromatin immunoprecipitation sequencing, ChIP-seq)是基因转录水平组蛋白修饰的最常用方法之一,该技术起初主要用于研究蛋白和DNA的相互作用。ChIP技术主要用于研究胞内相互作用和对研究基因水平的表观遗传学改变;ChIP-seq结合了ChIP和二代DNA测序技术,可以有效地检测特定组蛋白修饰和转录因子的DNA结合位点。ChIP-seq的原理为:DNA片段纯化与目标蛋白质结合并通过ChIP纯化富集和免疫共沉淀,然后纯化并构建文库,富集的DNA片段通过高通量技术进行测序;借助该技术,研究人员已经精确定位了基因组中与特定组蛋白相互作用的序列和转录因子。对于肿瘤微生物组研究而言,ChIP-seq技术适用于宿主和微生物细胞的基因组或表观遗传学研究。
2.5 三维基因组学
三维基因组学亦即空间基因组学,主要基于一维(1D)基因组序列的基本信息,研究基因组的三维结构和不同元件(包括转录因子和DNA/RNA结合蛋白)介导的转录调控机制。近年来,在该领域发展的技术主要包括高通量/高分辨率染色体构象捕获(high‐throughput/resolution chromosome conformation capture, Hi-C),基于微球菌核酸酶的染色体折叠分析(micrococcal nuclease‐based analysis of chromosome folding, Micro-C),使用长x-接头的基于微球菌核酸酶的染色体折叠分析(micrococcal nuclease‐based analysis of chromosome folding using long x‐linkers, Micro-C XL),通过成对末端标签测序的染色质相互作用分析(chromatin interaction analysis by paired‐end tag sequencing, ChIA-PET)和HiChIP(in situ Hi‐C followed by chromatin immunoprecipitation)。与肿瘤微生物组相关的研究主要集中于宿主细胞的真核基因组,最近Hi-C技术开始用于细菌染色体的研究,为生物样本DNA序列的物理邻近性提供依据。
2.6 基因组编辑
基因编辑也称为遗传修饰或基因工程,主要用生物化学的方法改变基因组序列,主要通过在基因组中插入目标片段或删除特定片段来实现。基因编辑历史悠久,常见工具包括转录激活因子样效应核酸酶(transcription activator‐like effector nucleases,TALENs)、成簇规律间隔短回文重复序列(clustered regularly interspaced short palindromic repeats, CRISPR)和CRISPR‐associated protein 9(Cas9)等。目前,CRISPR-Cas9技术及其衍生的技术是基因组编辑的主流技术。
肿瘤微生物组研究领域,基因编辑技术已广泛用于宿主和微生物,发现肠道微生物的基因组组成变化很大。单核苷酸多态性(SNPs)的系统发育分析显示了全球宏基因组样本中显著的种内遗传变异。因为我们可以看出CRISPR‐Cas9技术在菌群研究领域有较好的应用前景。
2.7 单细胞基因组学和转录组学
基因组和转录组学方法能提高覆盖率同时降低假阳性,但组装正确率取决于测序深度和群落复杂性,如宏基因组技术能研究复杂群体的细菌组成,但低丰度菌的序列可能被高丰度生物的序列掩盖。此外,具有相似表型的细胞中的遗传信息可能存在显著差异,且低丰度信息可能会丢失。单细胞测序技术弥补这些局限性的同时,可以揭示单个细胞的基因结构和基因表达,同时能更好地反映细胞间的异质性。
目前单细胞基因组和转录组主要基于4种研究方法:琼脂平板稀释、基于精细毛细管移液分离和倒置显微镜的单细胞外显子组测序、基于液滴的微流体技术和改进的荧光原位杂交(fluorescence in situ hybridization, FISH)法,而其中关键的一步是从组织或其他来源制备单细胞悬液。
2.7.1 琼脂平板稀释法
通过平板划线获得单一微生物的纯培养物,然后反复稀释微生物或不同细胞类型的混合物,从而将不同的群落分离成单细胞,这些单细胞将生长成同质菌落(图3A)。微流控划线平板(microfluidic streak plate, MSP)技术基于微流控的传统平板划线技术。MSP使用微流体得到微液滴,在培养皿上划线培养单细胞。传统琼脂平板稀释和MSP都可以整合到高通量筛选工作流程中。
2.7.2 基于倒置显微镜和口吸毛细管移液系统方法
组织样本的细胞悬浮液在倒置显微镜下用口吸毛细管系统或其他单细胞移液系统进行处理得到单细胞。分离的单细胞经确认后随机置于PCR管中,然后通过全基因组扩增(whole genome amplification, WGA)获得基因组序列信息,同时可用于外显子组测序(图3A)。这种技术已被用于研究肾细胞癌和邻近肾组织的研究,发现肿瘤的发生比以前认为的更复杂,并促进了更有效的细胞靶向治疗的发展。由此我们也可以断定,这种方法也可用于研究单个肿瘤细胞内的微生物的研究。
2.7.3 微滴微流体法
该方法主要用于基于两种不相容的液体来制备液滴,液滴的产生主要基于微管结构和两种液体之间的流速(图3A),固定流速的泵驱动两种液体进入不同的微通道,当它们相遇便产生微滴;这些液滴是理想的微反应室,可容纳单个细胞。单个液滴可通过系统进行填充、操作、分离、组合、检测和分类,从而得到数千个液滴。微滴微流体可以从大量细胞群中分离出单个细胞,通过一系列酶促步骤提取和分割基因组,遗传物质被扩增和标记后进行单细胞测序。该技术结合了微流体技术、DNA条形码和测序技术。
被包埋的微生物可以直接在液滴中培养,并在第一次扩增过程中生长上百个细胞,然后裂解纯化,用细胞分选仪将含有单细胞扩增DNA的珠子分选到标准PCR板中,随后再扩增到单细胞扩增基因组(single‐cell amplified genome, SAG)文库中。基于SAG-gel平台的典型单细胞测序方法(图3A)的微流控液滴发生器已用于在琼脂糖凝胶珠中培养小鼠肠道微生物。
包埋的单一微生物也可以直接裂解,然后进行DNA扩增或RNA逆转录和条形码标记。对于单细胞基因组测序,细胞通常首先被扩增,然后进行标记;单细胞转录组测序,通常首先标记细胞RNA,然后逆转录,最后是互补DNA(cDNA)扩增(图3A)。为了研究细胞内的核小体,核小体必须首先用条形码标记,然后进行免疫沉淀(single-cell ChIP-seq)和DNA扩增(图3A)。微滴微流体技术可以分析数百万个独立的反应,目前该技术也被用于单个DNA分子的深度测序,通过单细胞芯片测序和高通量分析对被标记的核小体进行分析。
微滴微流控单细胞测序无需单独培养微生物即可对细胞进行测序。通过这种方法,可以得到宏基因组数据库以研究肿瘤和其他区域(如肠道)中的微生物。这种方法的局限性在于,它从单拷贝基因组开始,但在酶和微流体处理过程中信息的损失不可避免,从而降低单个细胞信息的覆盖率。
2.7.4 改进的荧光原位杂交法
荧光原位杂交(FISH)是研究培养微生物的重要工具,可用于鉴定单细胞,也可用于标记靶向RNA(图3B)。RNA拷贝数可以反应在荧光水平上,因此该技术尤其适用于靶向rRNA的检测。16S rRNA不同区域的保守度不一样,可以根据菌类型设计特异性的引物。为解决分辨率低的问题,研究者们又开发了一些其他的方法,如催化报告分子沉积-荧光原位杂交(catalyzed reporter deposition‐FISH, CARD-FISH)使用辣根过氧化物酶(horseradish peroxidase, HRP)标记的探针以指数倍数放大信号,以便对单细胞进行特异性的荧光标记;Highly phylogenetic resolution FISH(HiPR‐FISH)技术在已有的单细胞自动图像分割的基础上执行像素分类和图像优化滤波。这些技术的发展对特定的微生物种群、含量低微生物和极低拷贝数RNA的可视化和分类及实现单细胞定量有重要作用。FISH可能会首先用于鉴别和定位肿瘤组织中的微生物。
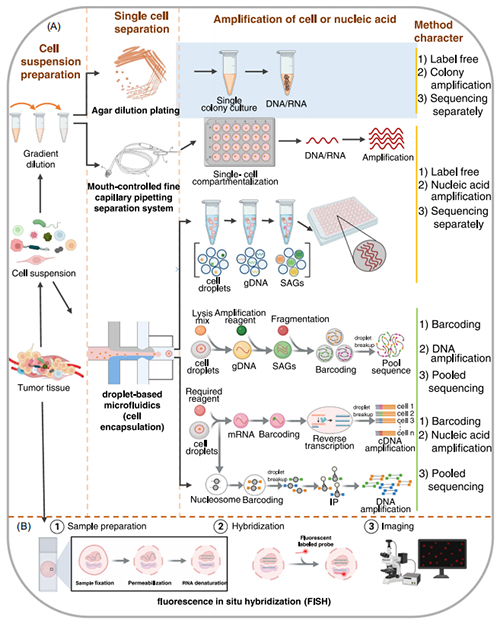
图3. 单细胞基因组和转录组方法
2.8 蛋白质组学
基因和转录的表达和调控水平可直接反应在成千上万的蛋白质的表达、PTMs和蛋白-蛋白间的相互作用水平上。液相色谱-质谱联用技术(Mass spectrometry coupled with liquid chromatography, LC-MS)是蛋白质研究中最常用的方法之一,且已成为肿瘤微生物组研究的有力工具。蛋白质组学研究有三种不同的策略:top‐down,middle‐down 和bottom‐up;其中top‐down(图4A)直接用MS对目标蛋白质进行检测,无需其他预处理;middle‐down首先将蛋白质酶解成大的肽段,然后用质谱进行检测;bottom‐up也称为鸟枪法,是使用最广的一种方法,蛋白质酶解成含有6-20个氨基酸的肽段(图4B)。
蛋白质组学的研究主要是筛选健康和疾病间的差异表达蛋白。目前基于质谱的蛋白质组学主要方法有数据依赖采集(data‐dependent acquisition,DDA)和数据非依赖性采集(data‐independent acquisition, DIA)(图4B)2种。在串联质谱(MS/MS)中,DDA只对部分母离子进行二级扫描;而DIA对所有母离子的MS2信息进行采集,因此DIA已成为临床样本(如肠道微生物样本)机制和生物标志物筛选研究中最受欢迎的数据采集方式,且DDA主要用于靶标检测。靶标蛋白质组学技术主要通过PRM(parallel reaction monitoring)或MRM(multiple reaction monitoring)方式对样本中的目标蛋白进行检测。蛋白定量包括相对定量和绝对定量,绝对定量首先通过标准曲线对样本中的蛋白进行定量;相对定量无需标准曲线,通过相对含量确定蛋白在2样本中差异倍数。
相对定量中,又有标记和非标记2种方法;非标记定量不需要同位素标记蛋白就可对不同生物样本中的蛋白质进行相对定量;标记定量主要有iTRAQ(isobaric tags for relative and absolute quantitation),TMT(tandem mass tag)和SILAC(stable isotope labeling by/with amino acids in cell culture),主要用同位素标签标记或者替换相应氨基酸;TMT(图4D)和iTRAQ主要用于体外标记,而SILAC用于体内标记(图4C)。标记的方法可将不同来源的蛋白的含量通过一次实验检测出来,且对低丰度蛋白和PTMs含量的改变有较好的灵敏度。
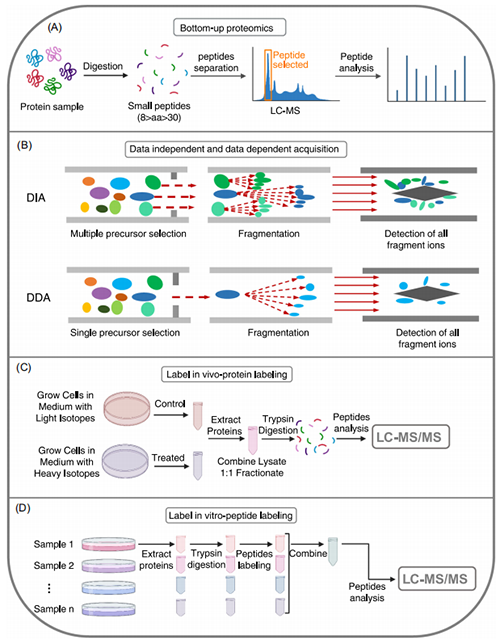
图4. 蛋白质组学的不同技术
2.9 代谢组学
代谢组学技术主要用于检测生物体内代谢物。代谢组学在肿瘤微生物组研究中致力于识别肿瘤微生物的代谢差异及其与周围环境的相互作用,能加深对肿瘤微生物影响肿瘤机制的理解。与微生物相关的代谢物主要包括短链脂肪酸(short‐chain fatty acids,SCFAs)、氨基酸类代谢物 、维生素、胆汁酸(bile acids, BAs)、乳酸、毒素和甲酸盐等。许多研究表明,微生物相关的代谢物可以正向或负向调控免疫、炎症、信号通路或改变肿瘤微生物(图5A),从而显著影响肿瘤的发生和发展。
SCFAs是肠道菌群的主要代谢物之一,可以对肠道功能和肠道代谢产生正向或负向的影响;另外已有研究发现补充某些产生SCFA的菌可以抑制肠道肿瘤的发展;此外,SCFAs也有望成为结直肠癌的诊疗靶点。异戊酸(Isovaleric acid, IVA)是一种与结直肠癌相关的肠道微生物相关的SCFA,可以促进蛋白质的表达进而导致下游5-羟色胺(5-HT)的增加;5-HT可直接作用于肿瘤干细胞,促进自我更新而增加肠道肿瘤发生。
微生物的其他代谢物也可以调节T细胞的代谢,如异石胆酸可以增强氧化磷酸化和线粒体ROS,促进调节性T细胞分化;一些回流BA可增加促进胃癌的发生的牛脱氧胆酸的产生。产毒素细菌艰难梭菌可激活肠上皮祖细胞中的信号通路,增加ROS产生和促进肿瘤的黏膜免疫反应;具核梭杆菌分泌的甲酸盐可以激活增加结直肠癌细胞的侵袭力相关的信号通路;来自肠道菌群的氧化三甲胺(Trimethylamine oxide, TMAO)增强CD8+T细胞介导的抗肿瘤免疫;没食子酸是肠道微生物的另一种代谢产物,也已被证明可以调节Treg细胞并增强癌症免疫治疗。肠道细菌及其代谢产物可通过诱导突变、增强肿瘤相关促炎反应、招募免疫抑制细胞等方式促瘤;肠道真菌还可以诱导促瘤炎症微环境,并产生真菌毒素从而诱导突变,促进肿瘤。
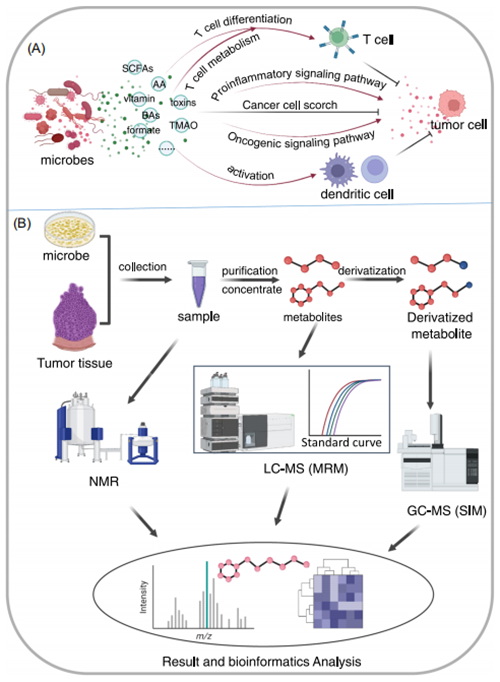
图5. 用于肿瘤微生物组研究的代谢方法
2.9.1 肿瘤微生物相关代谢物分析和样本预处理方法
代谢组学主要有NMR(nuclear magnetic resonance)和MS(mass spectroscopy)两个平台;NMR非破坏性的且不依赖于分离技术、样品制备相对较快、操作简便、成本低;向样品中加入已知浓度的内标化合物可以提高NMR的绝对定量;由于NMR灵敏度相对较低、微生物代谢物的复杂性以及代谢物浓度范围跨度大,从而限制了该平台在微生物代谢组学中的应用。
高分辨率MS成为代谢组学研究中的主流平台。利用包括LC、气相色谱(gas chromatography, GC)和毛细管电泳(capillary electrophoresis, CE)在内的分离技术克服了NMR平台存在的问题并广泛用于微生物代谢组学。GC-MS由于性能稳定且结果重现性好而广泛用于代谢组学研究,但GC-MS主要用于挥发性和热稳定性低分子量化合物的检测,而大多数微生物代谢物(如磷酸化化合物)是非挥发性的和热不稳定的代谢物,因此,对于微生物来源的代谢物需要经衍生化处理。与GC-MS和LC-MS相比,CE-MS用于代谢组学研究有较好的分辨率、所需样品量少且成本低,而CE和MS间的连接问题限制了其在代谢组学领域的应用。与其他技术平台相比,LC-MS和LC-MS/MS可以不经衍生化处理检测极性和高分子量物质,样品制备方便;此外,LC-MS灵敏度高,所需样品量少;此外,超高效液相色谱(ultrahigh performance liquid chromatography, UHPLC)的出现大大提高了LC-MS的色谱分辨率。
代谢组学主要分为靶向代谢组学和非靶向代谢组学;非靶向代谢组学对样本中的所有物质进行检测,而靶向代谢组学是指基于SRM和标准曲线对目标代谢物进行相对或绝对定量,TQMS(triple quadrupole MS)和传统的离子阱质谱(ion trap mass spectrometers, iTRAQ)是靶向代谢组学研究的主要技术平台。四极杆飞行时间(quadrupole time of flight, Q-TOF)质谱仪、傅里叶变换离子回旋共振(Fourier‐transform ion cyclotron resonance, FT-ICR)质谱仪和基于离子阱质量分析器轨道阱的质谱仪(Orbitrap)等高分辨率质谱仪,因采集速度快而广泛用于非靶向代谢组学研究。
鉴于胞内代谢随时间和外部环境快速变化,因此,取样和样品制备的方法和条件(如时间和储存条件等其他因素)对代谢检测的重现性、精密度和准确度有较大的影响。样品制备主要分为代谢淬灭和代谢物提取两个过程,淬灭是在尽量不损伤细胞膜的情况下快速停止胞内代谢;快速收集好的组织或细胞样品可快速置于液氮中淬灭,然后-80℃保存;此外,冷冻淬火和化学淬火或用酸或冷甲醇水溶液淬灭也是常用的淬灭方法。
样本中代谢物的提取可以通过物理方法,手工研磨或使用组织匀浆器实现,其中基于甲醇、氯仿和乙腈等的有机溶剂萃取由于提取效率高而常用于代谢物的提取,此外有机溶剂提取法对LC-MS的离子抑制效应也更小;基于NMR的分析方法无需对样本进行处理,样品制备简单。LC-MS和NMR在微生物相关代谢物检测分析方面各有其自身的优缺点,研究人员可以根据感兴趣化合物的特性和自身平台的特点来选择合适自己研究的方法。
2.10 单细胞蛋白质组学和代谢组学
单细胞测序主要用于细胞基因组、转录组和表观基因组的研究;同样,单细胞蛋白质组和单细胞代谢组在单细胞水平的基因组和转录组水平的研究中也有重要作用,但单细胞蛋白和代谢物的信号不能被放大;追踪是代谢组和蛋白组学研究中最大的挑战,因为这对于减少样品处理过程中细胞内分析物的损失至关重要。
单细胞蛋白质组学研究之前由于缺少明确和普遍适用的方法而只处于起步阶段;随着MS灵敏度的提高和样品制备方法的不断改进,单细胞蛋白和复合蛋白的绝对定量取得很大的进展。
基于Orbitrap的高分辨质谱在离子传输效率、检测灵敏度和分辨率等方面得到了极大的改善,尤其是最新一代的Orbitrap Eclipse MS上分析单个HeLa细胞研究发现,肽段和蛋白的覆盖率分别增加了36%和20%;最新的质谱仪器,如传统迁移谱(drift tube‐based ion mobility spectrometry, DT-IMS),捕获离子淌度质谱(trapped ion mobility spectrometry, TIMS)和高场非对称波形离子迁移谱(high-field asymmetric waveform ion mobility spectrometry, FAIMS),可以提高从多电核肽段选择性分离过滤单电荷肽段的能力,在非靶向蛋白质组中,单电荷肽段的滤除对于样本中蛋白的定性和定量有一定影响。
目前的单细胞代谢组学技术主要利用检测可培养微生物的代谢物组成,不能直接用于多细胞生物研究微生物与细胞间的相互作用。质谱成像(Mass spectrometry imaging, MSI)可以检测组织中的代谢物和其他生物分子,在单细胞代谢领域有巨大的应用前景。
基于MSI技术基质辅助激光解吸电离(Matrix‐assisted laser desorption/ionization, MALDI)质谱也是目前最常用的单细胞代谢组研究的分析技术;样品在进入质谱前与基质混合并用脉冲激光照射。MALDI-MS测量组织切片或载玻片上样本不同点的代谢物,从而创建代谢物的空间图谱;使用MALDI可以获得空间分辨率低于5μm的质谱图,这使得研究代谢物的分子分布更加可靠。与其他方法相比,MALDI-MS的样本前处理简单通量高,已用于单细胞生物体克隆群体中的异质性和组织中特异的微生物和细胞亚型研究;但MALDI存在代谢物覆盖率低和电离效率低的缺陷。为了克服MSLDI低覆盖率的缺陷,有科学家提出了一种基于不同电离度的多种基质的MSI方法,但该方法仍不能检出低离子能的代谢物。开发的MALDI-2通过在第二个激光器产生的气相激光羽流中启动额外的电离增强分析物信号。
MSI技术的改进对提高细胞和亚细胞水平上不同代谢物产生了很大的影响,但MSI仍存在许多挑战。MSI能获得已知和未知代谢物的信号,软件和数据库的改进大大提高代谢物鉴定准确性。
2.11 应用多组学方法进行肿瘤微生物组研究
单一组学研究不足以全面理解与肿瘤微生物相关的更复杂的生物学过程,基于多种技术平台的多组学技术成为强有力的研究工具。已有大量研究表明微生物产生的细胞毒性代谢物在肿瘤发生和发展过程中起重要作用,如糖酵解有氧和无氧途径的重要中间物质丙酮醛(methylglyoxal, MGO),也可以在甲基乙二醛合酶(methylglyoxal synthase)的作用下由微生物合成,MGO可以不通过酶解作用实现蛋白共价修饰,从而调节细胞代谢和转录相关的功能;此外,MGO可以通过与鸟嘌呤残基反应诱导DNA和RNA损伤,也能破坏有丝分裂期间破坏染色体分离直接影响基因组的完整性。此外,MGO还可以与蛋白的赖氨酸残基反应形成糖基化产物(advanced glycation end products, AGEs),进而影响细胞功能并导致炎症的发生。MGO作为一种糖化试剂,可导致代谢紊乱、表观遗传的改变和核小体不稳定;MGO还可以影响转录因子活性并改变基因转录,引起细胞应激因子的积累。组蛋白MGO糖基化的两阶段模型表明,低剂量的MGO可以通过促进转录而利于癌细胞的增殖,但过量的MGO可导致染色质交联,弱化转录并导致细胞死亡。
通过化学蛋白质组学技术的研究表明,高浓度MGO可通过诱导染色质中的组蛋白-组蛋白和组蛋白-DNA交联损伤其动力学特征;基于MGO糖基化的化学探针在MGO糖基化追踪、富集等方面有较好的应用前景,对潜在生化功能的理解也有一定的促进作用。由此可知,MGO能在基因组、转录组、蛋白质组和代谢组水平上调节肿瘤的发生和发展(图6),对MGO的研究也进一步表明,单一组学不足以全面了解这些蛋白质在生物学研究中的作用。
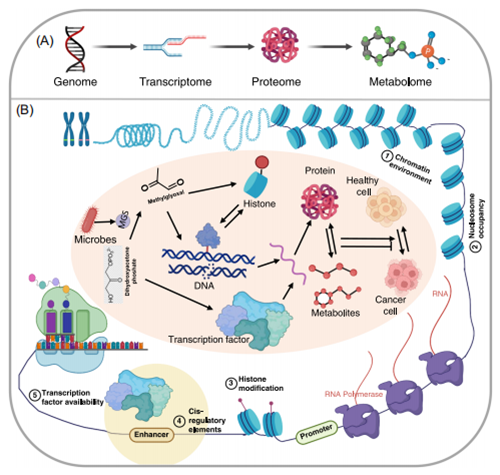
图6. 多组学应用促进相互关联学科的综合研究
3.未来展望
肿瘤微生物在肿瘤发生、发展和治疗过程中起着重要的作用;微生物、肿瘤细胞和免疫细胞之间的相互作用对肿瘤微环境有较大的影响。免疫细胞不仅影响微生物与抗癌疗法,而且能直接调节肿瘤发展过程中的微生物。因此,微生物治疗与免疫治疗相结合有望成为癌症治疗有效方法。多组学技术已经用于肿瘤微环境中的肿瘤细胞和微生物标志物研究,极大地促进了新诊断和治疗策略的发展。尽管组学技术在肿瘤领域取得了许多进展,但该领域的研究仍处于起步阶段,单细胞蛋白质组学和代谢组学技术在肿瘤微生物研究中很大程度上仍然是理论性的;此外,肿瘤微生物组多组学研究缺乏统一的标准流程。
本文系统总结了肿瘤微生物组研究的多组学方法及个方法的优缺点。尽管存在着肿瘤内微生物含量低且TME异质性高等问题,但单细胞多组学的发展必将克服这些难题,成为肿瘤微生物组研究中最有力的工具。
文献下载链接:
https://pan.baidu.com/s/1o1XJ_kn_Mv1Zl1vFjx-AMQ
提取码:0000







